
Dear Reader,
This is the second blogpost in a series which aims to explain the two most significant variational algorithms — VQE and QAOA. You can find the first part about VQE here — I will be referring to some concepts from the first blogpost, so please make sure you read it before starting this one.
In this article I will describe QAOA (Quantum Approximate Optimization Algorithm) — what’s the motivation behind it, how it works and what it’s good for.
We go pretty deep into details and motivation behind QAOA. If you have trouble fully understanding something — don’t worry. I’ve been using QAOA for solving different problems for 2 years and only recently started to really appreciate the math and physics behind it. You can always come back and revisit this blog post.
Table of contents:
- Combinatorial optimization problems
- Introduction to QAOA
- Background — AQC
- Background — Quantum Mechanics
- QAOA revisited
- What does the circuit look like?
- Why it’s appropriate for NISQ
- Relation to VQE
- Closing notes
One of the reasons why QAOA looks so promising is that perhaps it can help us solve combinatorial optimization problems better. Why should anyone care? Let’s see!
Combinatorial optimization problems
Evil imps sending notes in the classroom
Imagine a classroom where students are sending notes between each other. The teacher is annoyed by this and thanks to a meticulous invigilation knows exactly how many notes pass between each pair of students. The class is moving to a new room, where there is a huge gap in the middle, preventing students from sending notes to the other side. How should the teacher divide the class into two groups in order to minimize the number of notes being sent?
Or another example — in a far away kingdom, there were multiple merchants who all traded with each other. However, an evil imp wanted to impair the trade in this kingdom, so he decided to create an intrigue which would separate the merchants into two opposite factions, not trading between each other. The question was — who should be in which faction to maximize the losses?
What do both these situations have in common? They both can be described as a MaxCut problem — given a graph, find a way to divide it into two groups, such that the edges going between the two groups have the biggest possible weight.
In the first example, the nodes of the graph represent the students and the connections represent how many notes a given pair of students exchange. In the second example, the nodes are the merchants and the connections are the number of ducats, drachmas or talents their mutual trade is worth.
Let’s see what this graph might look like:
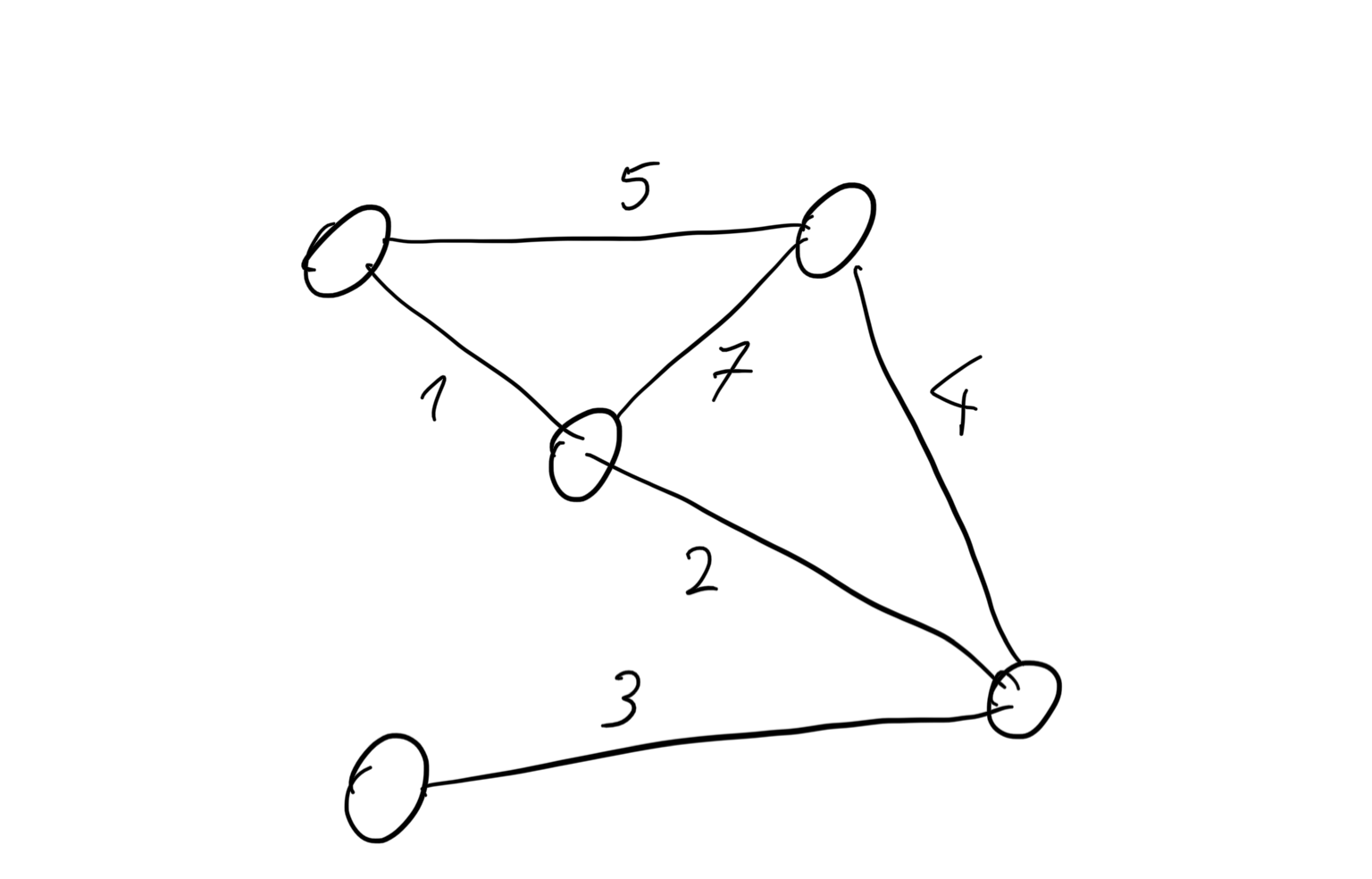
and what’s the solution of this problem:

Why MaxCut?
A small digression here: if you’ve seen your portion of graph and optimization problems, MaxCut might not be the most obvious example. However, we use it here for a few reasons:
- MaxCut is the problem that the authors of the original QAOA paper solved with this algorithm.
- It is the most well-studied example of QAOA application in the literature (probably because of 1).
- Solution to MaxCut is a binary string (so just a string of 0s and 1s) and there is no problem with encoding the results.
- As far as I can tell, it has some neat mathematical properties (especially for k-regular graphs) which make it easier to analyze. If you’re interested, you can find more answers in the original paper, as well as others papers by the same and these two talks: talk 1 and talk 2.
Why combinatorial optimization?
As you might have already guessed, MaxCut is an example of a combinatorial optimization problem (I’ll abbreviate it to COP). This is a class of problems where we want to find the best solution from a finite set of solutions — it usually involves graphs or ordering some objects. Some well-known examples are the Traveling Salesman Problem or the Knapsack Problem.
The first problem with combinatorial optimization is that it is, well, combinatorial. This usually means that the number of all possible solutions grows extremely fast with the size of the problem — so called “exponential growth” or “combinatorial explosion”. Exponential means that the number of possible solutions typically depends on \(a^n\) (\(a\) is a constant depending on a problem and \(n\) is the problem size). Combinatorial means that \(n!\) is involved — which is even worse than exponential.
That’s not a problem in and of itself — in the case of continuous optimization we have to deal with an infinite number of possible solutions (since we use real numbers) and somehow we have efficient methods of doing that. The fact that we need to deal with discrete values actually makes things harder — or at least different. Many continuous functions we have to deal with in optimization are smooth — i.e. if we change \(x\) just a little, the \(f(x)\) also changes just a little, which is really helpful. For discrete functions, if we change only one bit of the solution from 0 to 1, we might get dramatically different results.
It’s also worth noting that many such problems are NP-hard – for those unfamiliar with this term it basically means that there are no efficient methods for solving exactly big instances of a given problem. If you’re interested in details you can read on wikipedia, watch this video or read this book.
Since these problems are so difficult to solve, why do we bother? Because they are applicable in so many places that I won’t even try to list them here — apparently there is even a book just listing out where you can apply COPs.
QAOA — introduction
QAOA is an algorithm which draws from various concepts. We’ll start with a description of QAOA and then we’ll go one by one through all the pieces needed to actually understand why it works.
Let’s say we have a cost Hamiltonian \(H_C\) representing our problem (we will see how to create one in the section about Ising models). We can also construct an operator \(U(H_C, \gamma) = e^{-i \gamma H_C}\).
Then, we can construct another operator: \(H_B = \sum_{j=0}^n \sigma_j^x\) (\(\sigma_j^x\) is a pauli X matrix) and corresponding to it \(U(H_B, \beta) = e^{-i \beta H_B}\).
In QAOA we construct the state \(| \gamma, \beta \rangle = U(H_B, \beta_p) U(H_C, \gamma_p) … U(H_B, \beta_1) U(H_C, \gamma_1) | s \rangle\) , where \(p\) is usually called “number of steps” and denotes just how many times do we repeat applying \(U(H_B, \beta) U(H_C, \gamma)\). \(|s\rangle\) is the initial state, usually \(|0...0 \rangle\) or \(H | 0...0 \rangle = | +...+ \rangle\). We often call \(\beta\) and \(\gamma\) “angles”.
In order to find what are the values of angles which produce a reasonable state, we do exactly the same thing as we did with VQE. We start from some initial parameters, measure the state and update the angles to get a state closer to our solution in the next iteration.
The first time I looked at these equations I was like: “Ok, let’s implement it and see how it works!”. But the more I’ve been learning, the more I started to ask questions such as: “What do we need \(H_B\) for?”, “Why does \(H_B\) have this exact form?”, “Why do we use these funny \(e^{-iH}\) operators?” and, predominantly, “Why should it even work at all?”
Let’s find out the answers in the following paragraphs!
Background: Adiabatic Quantum Computation (AQC)
What’s AQC?
Let’s say we have a quantum system described with a simple Hamiltonian \(H_S\) with a known ground state. We also have another Hamiltonian \(H_C\), whose ground states we want to learn. We can now construct a new Hamiltonian in the form of: \(H(\alpha) = (1 - \alpha) H_S + \alpha H_C\). For \(\alpha=0\) our system is described with \(H_S\), for \(\alpha=1\) by \(H_C\).
The adiabatic theorem states that if we start in the ground state of \(H_S\) and very slowly start to increase \(\alpha\) up to 1, then throughout the process the system will always stay in the ground state of \(H(\alpha)\) — which means that at the end it will be in the ground state of \(H_C\). It’s also worth mentioning that \(\alpha\) could be an actual physical quantity.
Of course that’s not an easy thing to do and certain conditions apply, but in principle if the evolution is slow enough, we are able to find a solution to any problem if we can encode its solution in the ground state of \(H_C\). We use the word “evolution” for the process of changing the quantum system by changing \(\alpha\). Also, we often use \(\alpha=\frac{t}{T}\), where \(t\) is current time and \(T\) is total or end time.
Ising models
There is one big assumption in the previous paragraph, which could easily ruin any potential usefulness of this method: “if we can encode its solution in the ground state of \(H_C\).” Well, if we don’t know how to encode the problem, then AQC won’t help us. Fortunately, we know such encodings for many combinatorial optimization problems and we use “Ising models” to do that.
Imagine a chain of particles. Each particle has a spin which is either “up” or “down” (or +1 and -1) and it wants to have a different spin than its neighbours. We can write down the Hamiltonian for such system:
\[H(\sigma) = - \sum_{<i, j>} J_{ij} \sigma_i \sigma_j\]Here \(\sigma_i\) is the spin of the i-th particle and by \(\sigma\) we denote a string of all the spins — e.g. [-, -, +, + -, … ] — and \(J_{ij}\) is the strength of the interaction between the particles. This mathematical description actually works equally well with lattices instead of chains. Also, we can change the behaviour of the system by changing the sign of \(J_{ij}\) — if it’s negative it behaves as described, but if it’s positive, then the particles will try to have the same spin direction.
This model maps very well on quantum computers — we just need to change \(\sigma_i\) to \(\sigma_i^z\) (pauli Z operator acting on \(i\)-th qubit).
It turns out that we can encode many different COPs using Ising models — I won’t describe how to do that here, but you can find many examples in this paper. For our discussion it suffices to know that it is doable and that we can do that using only pauli Z operators.
Problems with AQC
Ok, so we can encode most problems of interest into Hamiltonians and we have a method which basically guarantees that we find a proper solution. So what’s the catch?
Well, there are a couple of them:
- In order to work AQC requires to be very well isolated from the outside world — which is really hard to do in practice.
- It might require a very long time to run.
- You won’t be able to run it on a gate-based quantum computer. Well, while it is possible in principle, it would required much better hardware than we have today and doesn’t seem to be practical in any foreseeable future
Basically, the harder your problem is, the more isolated your quantum system must be and the longer it has to run.
Relation to quantum annealing
This seems like a good place to mention quantum annealing (QA) — the paradigm that Canadian company D-Wave is following. The main idea behind QA is the same as for AQC — start from a ground state of an easy Hamiltonian and slowly evolve your state towards the ground state of the interesting Hamiltonian. What’s the difference? QA is an imperfect implementation of AQC which trades some of AQC’s power for easier implementation. Although not everybody might agree on this since the line between what the community call AQC versus QA is very blurry, basically we can say that QA is a heuristic algorithm which:
- Can be more flexible when it comes to the rate of change between two Hamiltonians (finite practical time which is on the order of microseconds in current devices) and
- Operates at finite temperature.
This comparison leaves a lot of subtleties out, but I think it’s a good first approximation to start thinking about it. If someone would like to learn more, these two publications might shed some light: Comparison of QAOA with QA and SA and AQC and QA: Theory and Practice.
Background: Quantum Mechanics
In this section I’d like to review two ideas which come directly from basic quantum mechanics — time evolution of a quantum system and trotterization.
Time evolution
There is an operation which is quite common in quantum computing — taking an operator \(A\) and creating another of the form \(U_A = e^{-itA}\). But why would you do that?
Well, it comes from the Schrödinger equation:
\[i \hbar \frac{d}{dt} | \Psi(t) \rangle = H | \Psi(t) \rangle\]What it says is that the change (in time) of the state of a quantum system described by a time-independent Hamiltonian \(H\) depends solely on the form of this Hamiltonian.
And if we solve the equation we see that this dependency is:
\[| \Psi(t) \rangle = e^{-iHt} | \Psi(0) \rangle\]What does this tell us? That if we take some state, \(| \Psi(0) \rangle\) and act on it (evolve it) with a Hamiltonian \(H\) for a period of time \(T\), we will get a state \(| \Psi(T)\rangle = e^{-iHT} | \Psi(0 \rangle\).
If this sounds abstract, imagine an electron orbiting the proton (hydrogen atom again!). This system is defined solely by the interaction between these two particles and their initial states and we know how to describe the interaction in the form of a Hamiltonian. So by solving the Schrödinger equation we are able to get a formula describing the state of the system after time T. And if we do the math, we know it will look like this: \(| \Psi(T) \rangle = e^{-iHT} | \Psi(0) \rangle\)
The key point to remember? Whenever you see an operator which looks like this \(e^{-iHt}\), you can understand it as an evolution of the quantum system described by the Hamiltonian \(H\).
Until this point we’ve made one silent assumption: we’ve assumed that the Hamiltonian we wanted to solve was time-independent. But when we take a look at the Hamiltonian we used for AQC (\(H(\alpha) = (1 - \alpha) H_S + \alpha H_C\)) and change \(\alpha\) to \(t\), we cannot pretend it’s time-independent. Depending on how complicated this time-dependency is, this might complicate the solution we’ve presented a little bit or a lot. In the latter case, the problem might be impossible to solve, so it’s reasonable to make some approximations.
Trotterization
Another concept important for understanding QAOA is “Trotterization”. Most of the time we want to find the ground state of a Hamiltonian which is too complicated for us to deal with directly — then it might be useful to approximate it. An example can be (surprise), a time-dependent Hamiltonian.
To help us grasp the idea we’ll get back to the “classical world”: imagine a curve. Let’s say this curve is described by some function.
We can approximate it by a piecewise linear function. The more “pieces” we use, the better approximation we get:
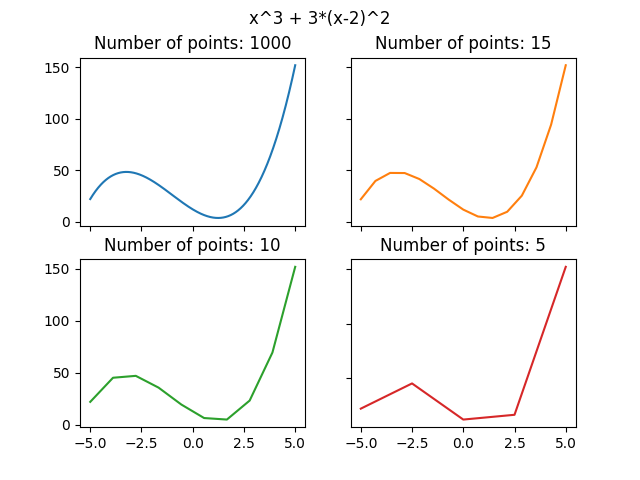
We can do the same with the time evolution of a quantum system for an operator \(U(t)\) (\(U(t)\) is actually a shorthand of \(U(t, t_0)\), which means that we act for time \(t\) starting from \(t_0\)). We can write \(U(t, t_0) = U(t_n, t_{n-1}) U(t_{n-1}, t_{n-2}) … U(t_2, t_1), U(t_1, t_0)\) (source). The bigger \(n\) we use, the better approximation we can get. Keep in mind that the time differences \(\Delta t_i = t_{i+1} - t_i\) don’t need to be equal.
Another approximation, particularly useful in quantum mechanics, is called the “Suzuki-Trotter expansion” or “Trotterization” (it is somewhat similar to other expansions we use in mathematics, like a Taylor series expansion. If you’re not familiar with this concept, I recommend watching this video by 3Blue1Brown ). If our Hamiltonian has the following form \(e^{A+B}\), we can use the following relation:
\[e^{A+B} = lim_{n \rightarrow \infty} (e^{\frac{A}{n}} e^{\frac{B}{n}} )^n\]We can look at it as approximating time evolution by \(A+B\) by applying alternatively \(A\) and \(B\) for time intervals \(\frac{1}{n}\). Now we have all the knowledge we need to understand what’s the motivation behind QAOA.
QAOA revisited
Now that we have some more context, let’s look at QAOA again.
QAOA creates the state: \(| \gamma, \beta \rangle = U(H_B, \beta_p) U(H_C, \gamma_p) … U(H_B, \beta_1) U(H_C, \gamma_1) | s \rangle\) , where \(U(H_B, \beta) = e^{-i \beta H_B}\) and \(U(H_C, \gamma) = e^{-i \gamma H_C}\) and \(|s\rangle\) is the starting state (we defined it before).
First, we can now see that the operators \(U_B = U(H_B, \beta)\) and \(U_C = U(H_C, \gamma)\) correspond to evolving the state with the Hamiltonian \(H_B\) and \(H_C\) for time \(\beta\) and \(\gamma\).
Second, we can also see that having several layers of \(U_B U_C\) looks a lot like a trotterization of \(e^{(H_B+H_C)}\). It’s not “pure” trotterization, since we have different \(\beta\) and \(\gamma\) for each step, so it resembles the “piecewise approximation”. But approximation of what?
Third, by combining the two previous points we can see that QAOA (somewhat) mimics adiabatic evolution from the ground state of \(H_B\) to the ground state of \(H_C\), which encodes our solution (remember Ising models?). So answering the question in the previous paragraph – QAOA could be (given right choice of parameters) viewed as the approximation of AQC.
This still leaves a couple of important questions open:
- What’s the actual relation between QAOA and AQC?
- Why should QAOA work?
- Why do we actually need \(H_B\)?
Relation to AQC
QAOA is not a perfect analogy to AQC. While there are a lot of similarities, there are also significant differences.
First of all, the goal is different. In AQC our goal is to get the ground state of the Hamiltonian. In QAOA (Quantum Approximate Optimization Algorithm) our goal is not really to get the exact ground state. It is to get a state which has good enough overlap with the ground state, so that we get an approximate solution.
Second of all, we could choose our angles in such ways that the evolution of a state using QAOA would mimic the evolution using AQC. But we don’t do that — in QAOA we can choose angles arbitrarily (usually using some classical optimization strategy).
And lastly, there’s something that I struggled a lot to understand (honestly, I still struggle with it ;) ). In AQC we start from \(H_B\) and slowly move towards \(H_C\). In QAOA we actually frantically alternate between \(H_B\) and \(H_C\) — more on that later.
Why should it work?
In principle, if you can get an infinite number of steps and choose angles which exactly mimic the adiabatic path, you will get the right results. It reminds me of the universal approximation theorem for neural networks. UAT says that if you have a one-layer neural network which is big enough, it can approximate any function, so basically solve any problem. The problem is with the “big enough” part — the requirements are astronomical for relatively simple problems. That’s why we usually don’t use gargantuan one-layer networks, but rather just stack smaller layers on top of each other. With QAOA the case is similar — in principle we could just keep increasing the number of steps and stay close to the parameters for the adiabatic path, but in practice, we choose not to. We try to solve the problem with a smaller number of steps by finding the right set of angles.
So we have some intuitions why it should work, but to be honest, we don’t know if it will work (at least without infinite resources) — it’s an important open question. Some of the researchers (me ;) ) are optimistic, because as it was with neural networks, they had very limited practical applications until some improvements made them an extremely useful and widely used tool. But we still don’t know whether this is the case with QAOA — solving a toy problem with an algorithm is different from solving a real problem with it.
Actually, there’s a quote illustrating it from a recent paper by two of the three authors of the original QAOA paper:
No one knows if a quantum computer running a quantum algorithm will be able to outperform a classical computer on a combinatorial search problem.
Why is \(H_B\) important?
\(H_C\) is easy — it corresponds to the problem that we want to solve and hence we know why it looks the way it looks. But what about \(H_B\)? Why do we choose it to be just \(\sum_i^N \sigma_i^x\) and not something else? Why do we even need it at all?
First, it doesn’t need to be just \(\sum_i^N \sigma_i^x\). We want it to be something that does not commute with \(H_C\) and this choice of \(H_B\) meets this requirement and is super easy to implement (after the exponentiation we get a layer of \(R_x\) gates). This discussion might help you better understand why commutation of operators is significant. Another way to look at it is that choice of \(H_B\) changes the expressibility of our ansatz — it allows us to reach states that we would otherwise never reach (remember the robot arm from the VQE post?).
Second, why do we need it at all? One part of the answer is that it is just a part of the setup that came from AQC and the way we apply it comes from Trotterization. Unfortunately it doesn’t give us a lot of intuition or reason why not to drop it altogether. For me it’s easier to understand this problem if we look at it from the perspective of not having \(H_B\) at all. So what would happen in such a case?
We would be just repetitively applying \(U_C\). But once we got into a state which is the eigenstate of \(H_C\) we wouldn’t get any further. This is basic linear algebra — if we apply an operator to its eigenvector, it can change its length, but not direction. The same applies if we had a \(H_B\) which commutes with \(H_C\). So we need this intermediate step of applying \(H_B\) which allows us to escape from the local minimum. How do we make sure we escape it? Well, that’s where the classical optimization loop is useful – we try to find the right values of the parameters \(\beta\) and \(\gamma\) which make it happen.
I came up with a metaphor which might be helpful to get some more intuition. Imagine you’re in a forest full of traps and you want to find a treasure. \(H_C\) allows you to feel the treasure and move towards it, while \(H_B\) provides you with a set of rules on how to move to avoid the traps. Without \(H_C\) you have no idea which direction to go and without \(H_B\) you cannot make any moves. This metaphor definitely has some limitations, but should work for starters :)
What does the circuit look like?
Ok, so let’s take a peek at how it looks in practice. The simple problem that we want to solve is Maxcut on a triangular graph (see the picture below). Two edges have weight 10 and one weight has weight 1.
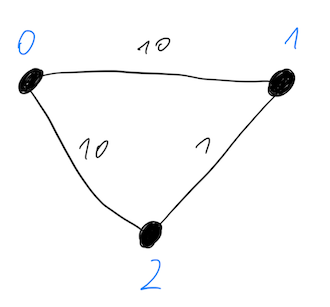
The correct solution to this problem is a cut which divides this graph into two groups: {0} and {1, 2}. We can encode solution into a binary string where 0 and 1 indicates in which group given node is, for example:
- 001: nodes 1 & 2 are in group 0, node 0 is in group 1
- 110: nodes 1 & 2 are in group 1, node 0 is in group 0
As you can see, we use representation where the rightmost bit is the least significant.
The cost function for MaxCut is the sum of costs of all the individual edges. Each individual cost is equal to: \(C_{ij} = \frac{1}{2} w_{ij} (1 - z_i z_j)\), where \(z_i=1\) if \(i-th\) node is in group 0 and \(z_i=-1\) if it’s in group 1. So if they’re both in the same group \(C_{ij}\) is equal to 0 and it’s 1 otherwise. I know such representation might not make a lot of sense at first glance, but once you start solving this problem you will see it’s reasonable. Now we can translate this into Hamiltonian form: \(\frac{1}{2} w_{ij} (1 - \sigma_i^z \sigma_j^z)\).
So in case of our graph, the Hamiltonian of interest is equal to:
\[H_C = \frac{1}{2} \cdot 10 \cdot (1 - \sigma_0^z \sigma_1^z) + \frac{1}{2} \cdot 10 \cdot (1 - \sigma_0^z \sigma_2^z) + \frac{1}{2} (1 - \sigma_1^z \sigma_2^z)\]Then we can take care of \(H_B\) — this is easy, it’s just \(\sigma^x_0 + \sigma^x_1 + \sigma^x_2\).
Now we need to build \(U_C\) and \(U_B\) from these and then create a circuit out of that. How?
Well, describing this procedure is beyond the scope of this blogpost. For those of you who are curious, you can find it in this paper, section 4. I have also not done this manually, I just used code from a qiskit tutorial. You can see the result below:

\(U_1 = e^{i \frac{\lambda}{2}} R_Z(\lambda)\) This is for \(\gamma = 0.5\) and \(\beta=0.3\).
This circuit consists of 4 parts:
- Hadamard gates — this is just for preparing the initial state — i.e. equal superposition of all states.
- \(U_C\) — in our graph we had 3 edges and we can see three blocks of gates here. Each consists of one two-qubit controlled \(U_1\) and two single qubit \(U_1\) gates. As you can see first two are parametrized with \(10 \gamma\), since the weights of the corresponding edges were equal to 10.
- \(U_B\) — this is just a layer of \(R_X\) gates parametrized with \(2 \beta\).
- Measurements
Why is it appropriate for NISQ?
If you don’t know what the issues with NISQ devices are, I described it already in the article about VQE. So here I’ll just point out why QAOA seems like a good algorithm for NISQ:
- The depth of the algorithm is scalable — the more steps we want to have, the more power QAOA has. With most current devices we’re limited to 1-2 steps (which are of little practical use), but this number will get higher with better hardware. Recently Google published results for \(p=3\).
- Similarly to VQE, QAOA is somewhat resistant to noise (which is also shown in the same paper from Google).
- Even though I’ve described only one particular way of creating the ansatz, there’s actually some flexibility and therefore you might be able to tune the algorithm toward specific architecture.
Relation to VQE
VQE and QAOA are often mentioned together. VQE is often described as an algorithm good for chemistry calculations and QAOA as good for combinatorial optimization. It’s worthwhile to understand the relation between the two.
QAOA can be viewed as a special case of VQE. In the end, the whole circuit we’ve described above is just an ansatz, parameterizable by the angles. And since the Hamiltonian contains only Z terms, we do not need to change the basis for measurements.
There are, however, a couple of things that make QAOA different:
- The form of the ansatz is limited to the alternating form we’ve already seen.
- We are restricted in terms of which types of Hamiltonians we can use (i.e. Ising Hamiltonians). This also means that we don’t need to do additional rotations before measurements as we did in VQE.
- The last (and I think the most fundamental) difference is the goal of the algorithm. In VQE we want to find the ground state energy and in order to do that we need to reproduce the ground state. In QAOA our goal is to find the solution to the problem. To do that we don’t need to find the ground state — we just need to find a state which has a high enough probability of finding the right solution.
Closing notes
Here are a couple of QAOA tutorials that you might find useful. I can’t say I’ve done them step by step, but I think they will be a good intro into how to use QAOA in practice:
- MaxCut tutorial by Guillame Verdon
- Various graphs tutorial by Jack Ceroni
- Qiskit QAOA tutorial
- My tutorial for Traveling Salesman Problem
As always I wanted to thank all the people that helped me make it both accurate and readable:
- Ntwali Bashige
- Borja Peropadre
- Alejandro Perdomo-Ortiz
- Yudong Cao
- Rishi Sreedhar
- Rafał Ociepa
If you want to learn more about variational quantum algorithms, you can read the next article in this series. And if you don’t want to miss the posts in this series about the state-of-the-art research on VQE and QAOA, please sign-up for the newsletter. You can do that using this link.
Have a nice day!
Michał